
Las dos glándulas suprarrenales, con un peso aproximado de 4 g cada una, se hallan en los polos superiores de los riñones. cada glándula se compone de dos porciones diferentes, la médula suprarrenal y la corteza suprarrenal. La médula suprarrenal, que ocupa el 20% central de la glándula, se relaciona desde el punto de vista funcional con el sistema nervioso simpático; secreta las hormonas adrenalina y noradrenalina en respuesta a la estimulación simpática. A su vez, estas hormonas provocan casi los mismos efectos que la estimulación directa de los nervios simpáticos en todas las regiones del cuerpo.
Corticoesteroides: mineralocorticoides, glucocorticoides y andrógenos
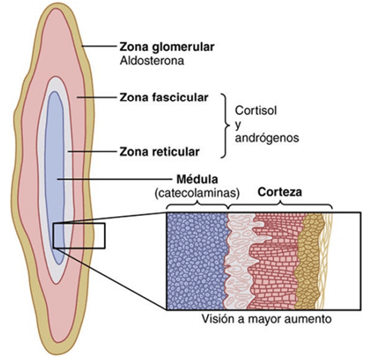
La corteza suprarrenal secreta los dos tipos principales de hormonas corticosuprarrenales, los mineralocorticoides y los glucocorticoides. Además de estas hormonas, produce pequeñas cantidades de hormonas sexuales, en particular de andrógenos, que inducen los mismos efectos que la hormona sexual masculina testosterona. En general, son de escasa importancia, pero cuando se secretan en grandes proporciones en algunos trastornos de la corteza suprarrenal (como se expondrá más adelante este capítulo), causan los efectos virilizantes consiguientes. Los mineralocorticoides reciben este nombre porque afectan sobre todo a los electrólitos los minerales del compartimiento extracelular, especialmente al sodio y al potasio. Los glucocorticoides se denominan así porque poseen efectos importantes de aumento de la glucemia. Además, influyen en el metabolismo de las proteínas y de los lípidos, con efectos tan importantes para la función del organismo como los que producen sobre el metabolismo de los hidratos de carbono. Se han aislado más de 30 esteroides de la corteza suprarrenal, pero tan solo dos son determinantes para la función endocrina normal del cuerpo humano: la aldosterona, que es el mineralocorticoide principal, y el cortisol, que es el glucocorticoide principal.
La corteza suprarrenal tiene tres capas diferentes:
- La zona glomerular, una capa delgada de células situada inmediatamente por debajo de la cápsula, contribuye con casi el 15% a la corteza suprarrenal. Estas células son las únicas de la glándula suprarrenal capaces de secretar cantidades importantes de aldosterona porque contienen la enzima aldosterona sintetasa, necesaria para la síntesis de la hormona. La secreción de estas células está controlada sobre todo por las concentraciones de angiotensina II y potasio en el líquido extracelular; ambos estimulan la secreción de aldosterona.
- La zona fascicular, la zona media y más ancha, representa casi el 75% de la corteza suprarrenal y secreta los glucocorticoides cortisol y corticosterona, así como pequeñas cantidades de andrógenos y estrógenos suprarrenales. La secreción de estas células está controlada, en gran parte, por el eje hipotalámico-hipofisario a través de la corticotropina (ACTH).
- La zona reticular, la capa más profunda de la corteza, secreta los andrógenos suprarrenales deshidroepiandrosterona (DHEA) y androstenodiona, así como pequeñas cantidades de estrógenos y algunos glucocorticoides. La ACTH también regula la secreción de estas células, aunque en ella pueden intervenir otros factores tales como la hormona corticótropa estimuladora de los andrógenos, liberada por la hipófisis. Sin embargo, los mecanismos que regulan la producción suprarrenal de andrógenos no se conocen tan bien como los de los glucocorticoides y mineralocorticoides.
Todas las hormonas esteroideas humanas, incluidas las producidas por la corteza suprarrenal, se sintetizan a partir del colesterol, Si bien las células de la corteza suprarrenal pueden sintetizar de novo pequeñas cantidades de colesterol a partir del acetato, casi el 80% del colesterol empleado para la síntesis de esteroides proviene de las lipoproteínas de baja densidad (LDL) del plasma circulante. Las LDL, que transportan altas concentraciones de colesterol, difunden desde el plasma al líquido intersticial para unirse a receptores específicos localizados en estructuras de la membrana de la célula corticosuprarrenal conocidas como depresiones revestidas. Estas depresiones penetran en el citoplasma por endocitosis, transformándose en vesículas que, por último, se fusionan con los lisosomas y liberan el colesterol destinado a la síntesis de los esteroides suprarrenales. El transporte del colesterol a las células suprarrenales está sometido a mecanismos de retroalimentación que pueden modificar en gran medida la cantidad disponible para la síntesis de esteroides. Por ejemplo, la ACTH, que estimula la síntesis de esteroides suprarrenales, incrementa el número de receptores de LDL de la célula corticosuprarrenal y la actividad de las enzimas que liberan el colesterol a partir de las LDL.
Etapas principales de la síntesis de los productos esteroideos más importantes de la corteza suprarrenal: aldosterona, cortisol y andrógenos.

Casi todas estas etapas suceden en dos orgánulos celulares, las mitocondrias y el retículo endoplásmico, pero algunas tienen lugar en las primeras y otras en el segundo. Cada etapa está catalizada por un sistema enzimático específico. Un cambio, incluso de una sola enzima, puede provocar la formación de tipos muy distintos y porcentajes diferentes de hormonas. Por ejemplo, si se altera la actividad de tan solo una enzima de esta vía, se generarán cantidades enormes de hormonas sexuales masculinizantes u otros compuestos esteroideos que normalmente no se encuentran en la sangre. Las fórmulas químicas de la aldosterona y el cortisol, que son las principales hormonas mineralocorticoide y glucocorticoide, respectivamente. la corteza suprarrenal suele secretar pequeñas cantidades de otros esteroides con actividad glucocorticoide, mineralocorticoide o mixta. Por último, se han sintetizado y empleado en diversas formas de tratamiento varias hormonas esteroideas potentes, no sintetizadas por las glándulas suprarrenales en condiciones normales.
- Mineralocorticoides
- Aldosterona (muy potente, supone casi el 90% de toda la actividad mineralocorticoide).
- Desoxicorticosterona (1/30 de la potencia de la aldosterona, aunque se secreta en cantidades mínimas).
- Corticosterona (ligera actividad mineralocorticoide).
- 9α-fluorocortisol (sintético, algo más potente que la aldosterona).
- Cortisol (actividad mineralocorticoide mínima, pero se secreta en grandes cantidades).
- Cortisona (actividad mineralocorticoide mínima). Glucocorticoides • Cortisol (muy potente; es el responsable de casi el 95% de toda la actividad glucocorticoide).
- Corticosterona (proporciona el 4% de la actividad glucocorticoide total, pero es mucho menos potente que el cortisol).
- Cortisona (casi tan potente como el cortisol).
- Prednisona (sintética, cuatro veces más potente que el cortisol).
- Metilprednisolona (sintética, cinco veces más potente que el cortisol).
- Dexametasona (sintética, 30 veces más potente que el cortisol).
Funciones de los mineralocorticoides: aldosterona La deficiencia de mineralocorticoides provoca pérdidas renales intensas de cloruro sódico e hiperpotasemia
La pérdida completa de la secreción corticosuprarrenal puede causar la muerte en un plazo de 3 días a 2 semanas, salvo que la persona reciba un tratamiento salino intensivo o la inyección de mineralocorticoides. Sin mineralocorticoides, la concentración del ion potasio del líquido extracelular experimenta un gran ascenso, el sodio y el cloruro desaparecen enseguida del organismo y el volumen total del líquido extracelular y el volumen de sangre se reducen mucho. Pronto se desarrolla un descenso del gasto cardíaco, que evoluciona a un estado de shock, seguido de la muerte. Toda esta secuencia puede evitarse con la administración de aldosterona u otro mineralocorticoide. En el ser humano, la aldosterona es la responsable de casi el 90% de la actividad mineralocorticoide de las secreciones corticosuprarrenales, pero el cortisol, el principal glucocorticoide secretado por la corteza suprarrenal, también aporta una actividad mineralocorticoide importante. La actividad mineralocorticoide de la aldosterona es alrededor de 3.000 veces mayor que la del cortisol, pero la concentración plasmática de este último es casi 2.000 veces superior a la de la aldosterona. El cortisol puede unirse asimismo a receptores mineralocorticoides con alta afinidad. Sin embargo, las células epiteliales renales expresan la enzima 11β-hidroxiesteroide deshidrogenasa de tipo 2 (11βHSD2), que tiene acciones que evitan que el cortisol active los receptores mineralocorticoides. Una acción de la 11β-HSD2 consiste en convertir el cortisol en cortisona, que no muestra avidez por unirse a receptores mineralocorticoides.
La aldosterona ejerce casi los mismos efectos sobre las glándulas sudoríparas y salivales que sobre los túbulos renales.
Estos dos tipos de glándulas producen una secreción primaria que contiene grandes cantidades de cloruro sódico, aunque gran parte del cloruro sódico se reabsorbe al atravesar los conductos excretores, mientras que los iones potasio y bicarbonato se excretan. La aldosterona aumenta de manera considerable la reabsorción de cloruro sódico y la secreción de potasio por los conductos. El efecto sobre las glándulas sudoríparas reviste interés para conservar la sal del organismo en ambientes cálidos y el efecto sobre las glándulas salivales permite conservar la sal cuando se pierden cantidades excesivas de saliva. Desde hace muchos años se conocen los efectos generales de los mineralocorticoides sobre el organismo, pero se ignora el mecanismo molecular de la acción de la aldosterona incrementa el transporte de sodio en las células tubulares. No obstante, la secuencia celular de acontecimientos que culmina con el aumento de la reabsorción de sodio parece ser la siguiente.
- En primer lugar, la aldosterona difunde de inmediato al interior de las células del epitelio tubular, debido a su liposolubilidad en las membranas celulares.
- En segundo lugar, la aldosterona se une a la proteína receptor mineralocorticoide (MR), una proteína que dispone de una configuración estereomolecular por la que solo la aldosterona o compuestos muy parecidos se unen a ella. Aunque los receptores MR de células epiteliales tubulares renales también poseen una alta afinidad por el cortisol, la enzima 11β-HSD2 convierte normalmente la mayor parte del cortisol en cortisona, que no se une fácilmente a los receptores MR, como se expone anteriormente.
- En tercer lugar, el complejo aldosterona-receptor o algún producto de este complejo difunde al interior del núcleo, donde sufre nuevas alteraciones para, por último, inducir la síntesis de uno o más tipos de ARN mensajero (ARNm) relacionados con el transporte del sodio y del potasio. En cuarto lugar, el ARNm pasa al citoplasma, donde, en colaboración con los ribosomas, induce la formación de proteínas.
Las proteínas así formadas consisten en:
- una o más enzimas
- proteínas transportadoras de membrana, cuya presencia conjunta es imprescindible para el transporte de sodio, potasio e hidrógeno a través de la membrana celular.
La regulación de la secreción de aldosterona está tan íntimamente ligada al control de las concentraciones de electrólitos en el líquido extracelular, el volumen del líquido extracelular, el volumen sanguíneo, la presión arterial y muchos aspectos especiales de la función renal que resulta difícil exponerla con independencia de todos ellos. Este tema se expuso con más detalle en los capítulos 29 y 30, a los que se remite al lector. Sin embargo, conviene enumerar aquí los aspectos más relevantes del control de la secreción de aldosterona. La regulación de la secreción de aldosterona por las células de la zona glomerular no depende apenas de la regulación del cortisol o de los andrógenos por las zonas fascicular y reticular.
Se conocen los siguientes cuatro factores que desempeñan una función esencial para la regulación de la aldosterona:
- El incremento de la concentración de iones potasio en el líquido extracelular aumenta mucho la secreción de aldosterona.
- El aumento de la concentración de angiotensina II en el líquido extracelular también incrementa mucho la secreción de aldosterona.
- El incremento de la concentración de iones sodio en el líquido extracelular apenas reduce la secreción de aldosterona.
- Se necesita ACTH de la adenohipófisis para que haya secreción de aldosterona, aunque su efecto regulador sobre la velocidad de secreción es mínimo en la mayoría de los trastornos fisiológicos.
Funciones de los glucocorticoides
Los mineralocorticoides pueden salvar la vida de los animales sometidos a suprarrenalectomía aguda, pero estos animales no se encuentran ni mucho menos bien. En realidad, los sistemas metabólicos animales de utilización de las proteínas, hidratos de carbono y lípidos están muy alterados. Además, el animal no resiste ningún tipo de estrés físico o mental y cualquier enfermedad leve, como una infección respiratoria, puede causar su muerte. Por tanto, los glucocorticoides ejercen funciones tan esenciales para prolongar la vida de los animales como las de los mineralocorticoides. Estas funciones se expondrán en los siguientes apartados. Al menos el 95% de la actividad glucocorticoide de las secreciones corticosuprarrenales se debe a la secreción de cortisol, también conocido como hidrocortisona.
Efectos del cortisol sobre el metabolismo de los hidratos de carbono
Estimulación de la gluconeogenia El efecto metabólico más conocido del cortisol y de otros glucocorticoides consiste en estimular la gluconeogenia (es decir, la formación de hidratos de carbono a partir de las proteínas y de otras sustancias) en el hígado; el ritmo de gluconeogenia se eleva, a menudo, entre 6 y 10 veces.
Este aumento del ritmo de la gluconeogenia se debe, sobre todo, a los efectos directos del cortisol en el hígado, así como a la antagonización de los efectos de la insulina.
- El cortisol aumenta las enzimas que convierten los aminoácidos en glucosa dentro de los hepatocitos. Este efecto se debe a la capacidad de los glucocorticoides para activar la transcripción del ADN en el núcleo del hepatocito, de la misma manera que la aldosterona actúa en las células del túbulo renal: se forman ARNm que, a su vez, dan origen al conjunto de las enzimas necesarias para la gluconeogenia.
- El cortisol moviliza los aminoácidos de los tejidos extrahepáticos, sobre todo del músculo. Por ello, llegan más aminoácidos al plasma, para incorporarse a la gluconeogenia hepática y facilitar la formación de glucosa.
- El cortisol antagoniza los efectos de la insulina para inhibir la gluconeogenia en el hígado.
El cortisol también reduce, aunque en grado moderado, la utilización de glucosa por la mayoría de las células del cuerpo. Aunque se desconoce la causa exacta de este descenso, un efecto importante del cortisol es la reducción de la translocación de los transportadores de glucosa GLUT-4 en la membrana celular, en especial en las células del músculo esquelético, lo que conduce a resistencia a la insulina. Los glucocorticoides también disminuyen la expresión y la fosforilación de otras cascadas de señalización que influyen en la utilización de la glucosa de forma directa o indirecta al afectar al metabolismo de las proteínas y los lípidos.
El incremento de la glucemia se debe tanto al incremento de la gluconeogenia como a la reducción moderada de la utilización celular de la glucosa. A su vez, el aumento de la concentración de glucosa estimula la secreción de insulina. Sin embargo, la elevación de los valores plasmáticos de insulina no resulta tan eficaz para mantener la glucosa plasmática como en condiciones normales. Por las razones que se expusieron anteriormente, los valores elevados de glucocorticoides reducen la sensibilidad de muchos tejidos, en particular del músculo esquelético y del tejido adiposo, a los efectos favorecedores de la captación y utilización de glucosa característicos de la insulina. Además de los posibles efectos directos de cortisol en la expresión de los transportadores de la glucosa y las enzimas que intervienen en la regulación de la glucosa, las altas concentraciones de ácidos grasos, causadas por el efecto movilizador de los lípidos de sus depósitos por los glucocorticoides, podrían alterar las acciones de la insulina sobre los tejidos. En consecuencia, el exceso de secreción de glucocorticoides provocaría anomalías del metabolismo de los hidratos de carbono, muy parecidas a las observadas en los pacientes con exceso de hormona del crecimiento. El incremento de la glucemia alcanza a veces tal proporción (50% o más sobre el límite normal) que se llega a un estado conocido como diabetes suprarrenal. En esta, la administración de insulina reduce la glucemia solo de manera moderada (no tanto como en la diabetes pancreática), porque los tejidos adquieren resistencia a los efectos de la hormona. Uno de los principales efectos del cortisol sobre los sistemas metabólicos del organismo consiste en el descenso de los depósitos de proteínas de la práctica totalidad de las células del organismo, con excepción de las del hígado. Esta reducción se debe tanto al descenso de la síntesis como a un mayor catabolismo de las proteínas ya existentes dentro de las células. Ambos efectos podrían achacarse a un menor transporte de los aminoácidos a los tejidos extrahepáticos, como se expondrá más adelante; es posible que esta no sea la causa primordial, porque el cortisol también reduce la formación de ARN y la síntesis posterior de proteínas de muchos tejidos extrahepáticos, sobre todo del músculo y del tejido linfático. Cuando existe un gran exceso de cortisol, el músculo puede debilitarse tanto que la persona es incapaz de alzarse cuando se encuentra en cuclillas. Además, las funciones inmunitarias del tejido linfático caen hasta una pequeña fracción de la normalidad.
El cortisol es importante para resistir el estrés y la inflamación
Prácticamente cualquier tipo de estrés, ya sea físico o neurógeno, provoca un aumento inmediato y notable de la secreción de ACTH por la adenohipófisis, seguido unos minutos después de una secreción considerable de cortisol por la corteza suprarrenal.
Efectos antiinflamatorios de las concentraciones altas de cortisol
Cuando un tejido sufre daños a causa de un traumatismo, una infección bacteriana o cualquier otra causa, suele inflamarse. A veces, como ocurre en la artritis reumatoide, la inflamación resulta más nociva que el traumatismo o la enfermedad. La administración de grandes cantidades de cortisol permite, de ordinario, bloquear esta inflamación o incluso revertir muchos de sus efectos, una vez iniciada. La inflamación consta de cinco etapas fundamentales:
- liberación por las células dañadas del tejido de sustancias químicas que activan el proceso inflamatorio, tales como histamina, bradicinina, enzimas proteolíticas, prostaglandinas y leucotrienos
- aumento del flujo sanguíneo en la zona inflamada, inducido por alguno de los productos liberados de los tejidos, un efecto que se denomina eritema
- salida de grandes cantidades de plasma casi puro desde los capilares hacia las zonas dañadas, secundaria a un aumento de la permeabilidad capilar, seguida de la coagulación del líquido tisular, con el consiguiente edema sin fóvea
- infiltración de la zona por leucocitos,
- crecimiento de tejido fibroso pasados unos días o semanas, para contribuir a la cicatrización.
Cuando se secretan o inyectan grandes cantidades de cortisol a una persona, el glucocorticoide ejerce dos efectos antiinflamatorios:
- puede bloquear las primeras etapas del proceso inflamatorio antes incluso de que se inicie una inflamación apreciable
- si la inflamación ya se ha iniciado, favorecerá su rápida desaparición y acelerará la cicatrización.
El cortisol ejerce los siguientes efectos preventivos de la inflamación:
- El cortisol estabiliza las membranas lisosómicas: esta estabilización es uno de los efectos antiinflamatorios de mayor interés, porque aumenta la resistencia a la rotura de las membranas de los lisosomas intracelulares. Por tanto, en las células dañadas se produce una importante disminución de la liberación de casi todas las enzimas proteolíticas que inducen la inflamación y que se encuentran normalmente en los lisosomas.
- El cortisol reduce la permeabilidad de los capilares, quizá como un efecto secundario a la menor liberación de las enzimas proteolíticas: este descenso de la permeabilidad impide la salida de plasma hacia los tejidos.
- El cortisol disminuye la migración de los leucocitos a la zona inflamada y la fagocitosis de las células dañadas. Sin duda, estas acciones se deben al descenso, inducido por el cortisol, de la síntesis de prostaglandinas y leucotrienos que, de otra manera, incrementarían la vasodilatación, la permeabilidad capilar y la movilidad de los leucocitos.
- El cortisol inhibe al sistema inmunitario y reduce mucho la multiplicación de los linfocitos, sobre todo de los linfocitos T. A su vez, la menor cantidad de linfocitos T y de anticuerpos en la zona inflamada amortiguan las reacciones tisulares que de otro modo fomentarían la inflamación.
- El cortisol disminuye la fiebre, sobre todo porque reduce la liberación de interleucina 1 por los leucocitos, uno de los principales estimuladores del sistema termorregulador hipotalámico. Por su parte, el descenso de la temperatura deprime la vasodilatación.
Regulación de la secreción de cortisol por la corticotropina procedente de la hipófisis
La ACTH estimula la secreción de cortisol A diferencia de la secreción de aldosterona en la zona glomerular, controlada sobre todo por el potasio y la angiotensina II que actúan directamente sobre las células de la corteza suprarrenal, la secreción de cortisol está sometida de forma casi exclusiva al control de la ACTH hipofisaria. Esta hormona, llamada también corticotropina o adrenocorticotropina, estimula asimismo la síntesis suprarrenal de andrógenos. Química de la ACTH La ACTH se ha aislado de forma pura de la adenohipófisis. Es un polipéptido grande, correspondiente a una cadena de 39 aminoácidos. Se conoce un polipéptido menor, un producto de la digestión de la ACTH, cuya cadena mide 24 aminoácidos pero que posee todos los efectos de la molécula entera.
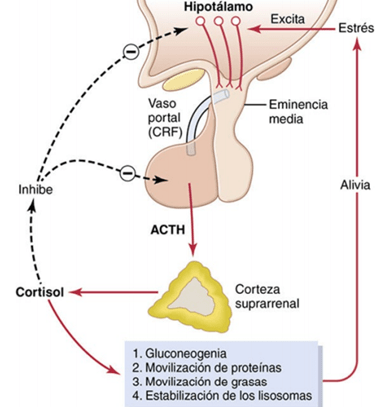
De manera idéntica al control de otras hormonas hipofisarias por los factores liberadores del hipotálamo, un factor liberador importante controla la liberación de ACTH. Se llama corticoliberina o factor liberador de corticotropina (CRF). Se secreta hacia el plexo capilar primario del sistema hipofisario portal en la eminencia media del hipotálamo y luego se transporta a la adenohipófisis, donde induce la secreción de ACTH. El CRF es un péptido formado por 41 aminoácidos. Los cuerpos celulares de las neuronas secretoras de CRF se localizan sobre todo en el núcleo paraventricular del hipotálamo. El efecto principal de la ACTH sobre las células corticosuprarrenales consiste en la activación de la adenilato ciclasa de la membrana celular. Esta activación, a su vez, induce la formación de AMPc en el citoplasma; el efecto máximo se alcanza a los 3 min. Por su parte, el AMPc activa las enzimas intracelulares que sintetizan las hormonas corticosuprarrenales. Se trata de un ejemplo adicional de la actuación del AMPc como segundo mensajero hormonal. El paso más importante de todos los estimulados por la ACTH para controlar la secreción corticosuprarrenal es la activación de la enzima proteína cinasa A, de la que depende la conversión inicial de colesterol en pregnenolona. Esta conversión inicial representa el paso limitante de la velocidad de síntesis de todas las hormonas corticosuprarrenales y explica por qué la ACTH se necesita, en condiciones normales, para la producción de cualquier hormona corticosuprarrenal. La estimulación a largo plazo de la corteza suprarrenal por la ACTH no solo eleva la actividad secretora, sino que también causa la hipertrofia y proliferación de las células de la corteza, sobre todo de las de las zonas fascicular y reticular, donde se secretan el cortisol y los andrógenos.
El cortisol ejerce un efecto directo de retroalimentación negativa sobre:
- el hipotálamo, disminuyendo la síntesis de CRF
- la adenohipófisis, reduciendo la formación de ACTH. Ambos efectos retroactivos ayudan a controlar la concentración plasmática de cortisol.
Anomalías de la secreción corticosuprarrenal
Hipofunción corticosuprarrenal (insuficiencia corticosuprarrenal): enfermedad de Addison La enfermedad de Addison se debe a la incapacidad de la corteza suprarrenal para fabricar suficientes hormonas corticales; a su vez, en un elevado número de casos, la causa obedece a una atrofia o lesión primaria de la corteza suprarrenal. Esta atrofia se debe casi en el 80% de las ocasiones a un fenómeno de autoinmunidad dirigido contra la corteza suprarrenal. La hipofunción de las glándulas suprarrenales puede ocurrir también por destrucción tuberculosa o por la invasión de la corteza por un tumor maligno. En algunos casos, la insuficiencia suprarrenal es secundaria a un deterioro en la función de la hipófisis, que no consigue producir suficiente ACTH. Cuando la producción de ACTH es demasiado baja, la de cortisol y aldosterona disminuye y, finalmente, las glándulas suprarrenales pueden llegar a atrofiarse debido a la ausencia de estimulación de ACTH. La insuficiencia suprarrenal secundaria es mucho más común que la enfermedad de Addison, que en ocasiones se denomina insuficiencia suprarrenal primaria. En las siguientes secciones se describen las alteraciones que se observan en la insuficiencia suprarrenal primaria.
Deficiencia de mineralocorticoides
La falta de secreción de aldosterona reduce mucho la reabsorción de sodio por el túbulo renal y, en consecuencia, permite la pérdida de grandes cantidades de agua y de iones sodio y cloruro por la orina. El resultado neto es un descenso llamativo del volumen extracelular. Además, aparecen hiponatremia, hiperpotasemia y acidosis leve por ausencia de secreción de los iones potasio e iones hidrógeno que normalmente se intercambian por el sodio cuando este se reabsorbe. Como el líquido extracelular se reduce, el volumen plasmático disminuye y la concentración de eritrocitos aumenta de manera espectacular; el gasto cardíaco y la presión arterial también se reducen y, en ausencia de tratamiento, el paciente fallece por shock entre 4 días y 2 semanas después de que cese completamente la secreción de mineralocorticoides. Deficiencia de glucocorticoides El paciente con enfermedad de Addison no puede mantener la glucemia normal entre las comidas, porque la falta de secreción de cortisol hace que no pueda sintetizar cantidades importantes de glucosa a través de la gluconeogenia. Además, la ausencia de cortisol reduce la movilización de las proteínas y las grasas de los tejidos, por lo que también se deprimen otras muchas funciones metabólicas. Esta pereza de la movilización energética ante la falta de cortisol es uno de los efectos más perjudiciales de la deficiencia de glucocorticoides. Incluso aunque la persona disponga de cantidades excesivas de glucosa y de otros nutrientes, sus músculos se debilitarán, lo que indica que los glucocorticoides son necesarios para mantener otras funciones metabólicas de los tejidos, aparte del metabolismo energético. La falta de secreción adecuada de glucocorticoides también aumenta la sensibilidad de los enfermos de Addison a los efectos nocivos de los distintos tipos de estrés; de hecho, una infección respiratoria leve puede causar la muerte. Pigmentación melánica Otra característica de casi todos los pacientes con enfermedad de Addison es la pigmentación melánica de las mucosas y de la piel. La melanina no siempre se deposita de manera homogénea y puede producir manchas, sobre todo en las zonas de piel fina, como las mucosas de los labios o la delgada piel de los pezones. Se cree que el depósito de melanina obedece a este mecanismo: cuando disminuye la secreción de cortisol, se reduce también el mecanismo normal de retroalimentación negativa sobre el hipotálamo y la adenohipófisis, con lo que se produce una enorme liberación de ACTH y también de MSH. Es probable que sean estas cantidades elevadísimas de ACTH las responsables de casi todo el efecto pigmentario, porque estimulan la formación de melanina por los melanocitos de la misma manera que lo hace la MSH.
Hiperfunción corticosuprarrenal: síndrome de Cushing
La hipersecreción corticosuprarrenal provoca una cascada compleja de efectos hormonales, conocida como síndrome de Cushing. Muchas anomalías de este síndrome se deben al exceso de cortisol, aunque la secreción exagerada de andrógenos también ocasiona efectos importantes.
El hipercortisolismo obedece a múltiples causas, por ejemplo:
- adenomas adenohipofisarios secretores de grandes cantidades de ACTH que, a su vez, causan hiperplasia suprarrenal y exceso de cortisol
- anomalías de la función del hipotálamo que ocasionan un aumento de liberación de la hormona liberadora de corticotropina, con el consiguiente estímulo exagerado de la secreción de ACTH
- secreción ectópica de ACTH por un tumor de otra parte del cuerpo, como un carcinoma abdominal
- adenomas de la corteza suprarrenal. Si el síndrome de Cushing es secundario a una secreción excesiva de ACTH por la adenohipófisis, el cuadro recibirá el nombre de enfermedad de Cushing. La causa más frecuente del síndrome de Cushing, que se caracteriza por un incremento de los valores plasmáticos de ACTH y de cortisol, es una secreción exagerada de ACTH.
La hiperproducción primaria de cortisol por las glándulas suprarrenales justifica entre el 20 y el 25% de los casos clínicos del síndrome y suele ir acompañada de un descenso de la ACTH, debido a la inhibición por retroalimentación de la secreción adenohipofisaria de esta por el cortisol. La administración de grandes dosis de dexametasona, un glucocorticoide sintético, permite diferenciar entre el síndrome de Cushing dependiente de la ACTH y la forma independiente de la ACTH. Normalmente, las dosis bajas de dexametasona no suprimen la secreción de ACTH de los pacientes con hiperproducción de hormona por un adenoma hipofisario secretor de ACTH o por una disfunción del eje hipotalámico-hipofisario. Al aumentar la dosis de dexametasona a niveles muy elevados, la ACTH termina por ser suprimida en la mayoría de los pacientes con enfermedad de Cushing. El tratamiento del síndrome de Cushing consiste en extirpar el tumor suprarrenal, si esta es la causa del proceso, o reducir la secreción de ACTH, si es posible. La hipertrofia de la hipófisis o incluso los microadenomas hipofisarios secretores de ACTH pueden extirparse mediante cirugía o se destruyen con radiación en algunos casos. Los medicamentos que bloquean la esteroidogenia, tales como metirapona, ketoconazol y aminoglutetimida, o que inhiben la secreción de ACTH, como los inhibidores de la GABA-transaminasa y los antagonistas de la serotonina, también se emplean cuando la cirugía no es posible.
Hiperaldosteronismo primario (síndrome de Conn)
A veces se desarrolla un pequeño tumor en la zona glomerular y se produce una gran secreción de aldosterona; el estado resultante se conoce como hiperaldosteronismo primario o síndrome de Conn. Asimismo, en casos aislados, la corteza suprarrenal hiperplásica secreta aldosterona y no cortisol. Los efectos del exceso de aldosterona se expusieron ya con detalle en este mismo capítulo. Las consecuencias más importantes son hipopotasemia, alcalosis metabólica leve, un ligero aumento del volumen extracelular y del volumen sanguíneo, un incremento mínimo de la concentración plasmática de sodio. S
Síndrome adrenogenital
En ocasiones, un tumor de la corteza suprarrenal secreta cantidades exageradas de andrógenos, que provocan efectos virilizantes intensos. Si este fenómeno afecta a una mujer, esta desarrollará características masculinas, como crecimiento de la barba, voz con un tono más grave de la voz o incluso calvicie, si es portadora del rasgo genético para la calvicie, distribución masculina del vello corporal y púbico, crecimiento del clítoris hasta parecerse al pene y depósito de proteínas en la piel y, sobre todo, en los músculos, con un aspecto típico masculino. Los tumores suprarrenales virilizantes tienen el mismo efecto en los varones prepuberales que en el sexo femenino, aunque aceleran el crecimiento de los órganos sexuales masculinos correspondiente a un niño de 4 años con síndrome adrenogenital. Las propiedades virilizantes del síndrome adrenogenital en los varones adultos suelen resultar enmascaradas por las características virilizantes habituales de la testosterona secretada en los testículos.
